
Today we cover the Form 483 [What is a Form 483?] issued by the US FDA [United States Food and Drug Administration] to Dr. Reddy’s oncology formulation [Duvvuda unit makes cytotoxic and hormonal injectables] manufacturing facility at Duvvada at Visakhapatnam, Andhra Pradesh.
On the same note, you can catch the other Form 483 issued to the API manufacturing plant at Miryalaguda here.
![]()
You can catch all our detailed coverage of Form 483s issued here for Alkem Laboratories, Divi’s Laboratories, Lupin, Glenmark and Sun Pharmaceuticals. Additionally you can catch all content written on pharmaceuticals here.
This is not a recommendation to buy or sell the stock. Analyst and family do own some of the positions listed here. Please assume we are biased.
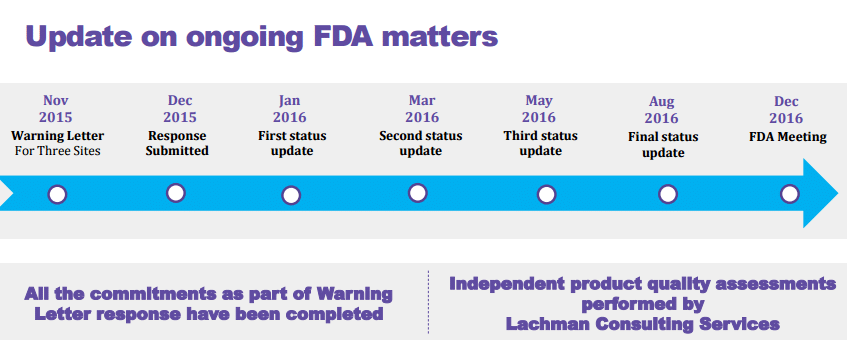
On the 8th March, 2017 – the US FDA completed their inspection of the company’s oncology formulation manufacturing facility at Duvvada, Visakhapatnam. The FDA inspection team reported 13 observations which was confirmed by the company’s in its press release to the regulatory body.

This re-inspection followed the earlier inspections of these sites by the agency in November 2014, January 2015 and February 2015 respectively. The company had provided multiple status updates to the FDA team post which this re-inspection took place.
The warning letter [in 2015] upset the company’s scheduled launch of the product Imatinib [key blood cancer treatment drug the generic version of Novartis blockbuster drug Gleevec which was due to launch in the fourth quarter of FY17 and has close to $1.5-2 billion sales in US with just four players] which was due to be launched as per the deal (settlement).
While the company went ahead and shifted the manufacturing [the company has earmarked about $40 million investment as part of remediation costs to be spent over an year and address the quality issues] from this oncology site that received a warning letter, it so happened that the partner site wherein it shifted the product to also went through the FDA audit raising further questions. While the company did not disclose the other products that may be affected, it did confirm one other injectable product that is a little behind from a launch along with other smaller products [not much of an impact].
 This is what the company had mentioned about the regulatory issues during the third quarter earnings call:
This is what the company had mentioned about the regulatory issues during the third quarter earnings call:
On the quality front, as communicated earlier, our warning letters impacted sites are scheduled to get re-audited during the months of February and March. A substantial remediation work has been put in place from our side. Our application of Corrective and Preventive Actions or CAPAs were not just site specifics, but they were also network wide and incorporated third party review and assessments. We believe we have prepared ourselves well for the audit. In the process of implementing the CAPAs, we have made significant progress in enhancing our quality systems and instilling the culture of quality and continuous improvement.
A quick flashback on the observations noted by the team which led to the issuance of the warning letter. Catch the complete letter here.
- Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed. (21 CFR 211.192).We note that the lack of adequate investigations is a repeat violation from our February 2008 inspection.In response to this letter, provide your plan to improve your investigations of critical process deviations. Include the changes you will implement to ensure that media fills provide adequate simulations. Detail how the critical interventions that may compromise the sterility of a product will be minimized in your aseptic processing operation.
- Your firm failed to follow appropriate written procedures designed to prevent microbiological contamination of drug products purporting to be sterile, and that include validation of all aseptic and sterilization processes (21 CFR 211.113(b)).The lack of appropriate written procedures designed to prevent microbiological contamination of drug products purporting to be sterile is a repeat violation from our February 2008 inspection.
- Your firm failed to establish adequate written procedures for production and process controls designed to assure that the drug products you manufacture have the identity, strength, quality, and purity they purport or are represented to possess, and your firm’s quality control unit did not review and approve those procedures, including any changes. (21 CFR 211.100(a)).In your March 27, 2015 response, you stated that you will create a protocol for preparing new qualification kits and documenting employees’ qualifications. Your response is inadequate. You did not indicate how previously inspected products may have been affected by your substandard visual inspection procedures and qualification kits. You also failed to provide the new protocol. Additionally, you did not provide any details on how you intend to train and qualify operators or measure the effectiveness of the new qualification kits.
Now, lets move ahead covering the latest inspection which resulted in 13 observations from the previously 3 observations.
 Ob1: There is a failure to thoroughly review any unexplained discrepancy whether or not the batch has already been distributed.
Ob1: There is a failure to thoroughly review any unexplained discrepancy whether or not the batch has already been distributed.
Ob2: Written procedures for production and process controls designed to assure that drug products have the identity, strength, quality and purity they purport or are represented to posses have not been established and followed.
Ob3: Failure to maintain complete data to ensure compliance with established specifications and standards.
Ob4: Production records do not contain complete and accurate information.
Ob5: Written procedures designed to prevent microbiological contamination of drug products purporting to be sterile are not followed, including validation of all aseptic process.
Ob6: Aseptic processing areas are deficient regarding the system for monitoring environmental conditions.
Ob7: Procedures for the preparation of master production and control records are not followed.
Ob8: Appropriate controls are not exercised over computer or related systems to assure that changes to master production records and control records or other records are instituted only by authorized personnel.
Ob9: Data is not documented contemporaneously.
Ob10: Thorough review of documents is not performed.
Ob11: Procedures for maintenance of equipment had not been established and followed.
Ob12: Samples collected for finished product release testing is not satisfactorily representative of the manufactured batch.
Ob13: Samples collected to evaluate conformance of a batch are not representative.
Here are the observations in detail. These can be our interpretation of the observations in general terms or replicated from the Form 483 if they are too technical in nature:
Ob1: There is a failure to thoroughly review any unexplained discrepancy whether or not the batch has already been distributed.
a. On the 27th February, 2017 – the FDA investigators noticed leakage in the filling operation. While an investigation were performed, the conclusion arrived was that the leakage was prior to the start of the filling process. Previous investigations into leakage incidents that occurred during manufacturing have not been thoroughly investigated to ensure root causes have been identified and appropriate corrective actions are implemented.
Despite repeated investigations, these incidents continue to occur which has been noted in multiple incident reports.
b. Collection of trending data for documentation errors, such as GDP errors, calculation errors, missing signatures, or incomplete documentation, began in May 2016. No critical evaluation of this data was performed to evaluate root causes of these errors.
224 errors in 17 Batch Manufacturing Records in July 2016, 128 errors in 21 Batch Manufacturing Records in August 2016, 143 errors in 22 Batch Manufacturing Records in September 2016, 200 errors in 21 Batch Manufacturing Records in October 2016.
No evaluation was performed to determine root causes of evaluate why localized training of the affected personnel was ineffective in eliminating errors.
c. Investigations into observations of objectionable organisms were not thorough
Burkholderia cepacia [aerobic gram-negative bacillus found in various aquatic environments], Vibrio vulnificus [same family as bacteria that cause cholera] and Acinetobacter baumannii [opportunistic bacterial pathogen primarily associated with hospital-acquired infection] were found in the samples tested in 2014. However, no root cause was determined and no corrective actions were performed though sanitization was performed and repeated again.
d. Staphylococcus aureus [group of bacteria that can cause a multitude of disease] was identified during active air monitoring in January, 2016. The investigation identified the likely root cause as improper gowning and hygiene of employees. Though the training was not held until March, 2016 while some were trained only in April, 2016 – there was no follow up done so as to check for the effectiveness of this training.
e. Equipment was not replaced after the integrity test failed twice, superior was not notified per instructions in SOP, and investigations were not initiated.

f. No assurance that the CAPAs are effective in addressing the actual causes of though the incidences attributed to analyst errors still continue.
g. Failure to identify the characteristics of the fiber/ plastic rejects or determine the source of fibers/ particulates in injectable products.
Ob2: Written procedures for production and process controls designed to assure that drug products have the identity, strength, quality and purity they purport or are represented to posses have not been established and followed.
a. The defect library used to train the visual inspectors did not include any examples of “black particles” for products until the 9th February, 2017.
This was a repeat observation from the 2015 warning letter – unacceptable procedures for qualification of visual inspectors.
Ob3: Failure to maintain complete data to ensure compliance with established specifications and standards.
Data integrity issue reported at another site of Dr. Reddy’s – injections included samples identified as “Blank” and “systemsuitability”. There was no incident investigation initiated for the this test and there is no explanation in the analytical records for the unreported sequence. Investigation and retrospective review for data integrity was not extended to the Chromeleon chromatography data generated at this site.
Video recordings for media fill batches have been destroyed though they are required to be made per the media fill protocols and were to be reviewed by QA.
Ob4: Production records do not contain complete and accurate information.
a. Biometric Entry Data which uses fingerprint technology to track and grant employees access to the facility indicated that personnel involved in the production process were not present during the actual production.
False and misleading statements were provided by employees on raising these observations.
b. Gowns inspection records show that critical area gowns rejected on the 23 February, 2017 were inspected and found acceptable on 27 and 28 February though the same gown was found in the waste area bag.
c. Records were signed and back dated by individuals who had not performed the steps and were not present during the process.
Ob5: Written procedures designed to prevent microbiological contamination of drug products purporting to be sterile are not followed, including validation of all aseptic process.
a. Requirements of what activities need to be performed during a media fill activity to qualify a person to person aseptic activities have not been established.
b. No entrance of exit logs to show which operators were present and when they present in the filling room.
c. Operation of the Online Continuous Particle Monitoring System does not describe actions to take when the non-viable particle count exceeds during set-up. Though the alarm occurred on the 7th March, the personnel performing the activities did not stop working [just prior to aseptic connections].
Ob6: Aseptic processing areas are deficient regarding the system for monitoring environmental conditions.
Microbiology media used to settle plates, active air samples and touch plate monitoring of the gloves do not contain neutralizing agents.
Ob7: Procedures for the preparation of master production and control records are not followed.
a. Master copies of raw data forms are available on a shared computer drive in the microbiology lab which can be modified with batch information and printed.
b. Blank GMP forms can be copied by laboratory of production personnel.
c. No effective way to ensure reconciliation of documents. QA does not reconcile the forms issued and returned if no additional pages were re-issued.
Ob8: Appropriate controls are not exercised over computer or related systems to assure that changes to master production records and control records or other records are instituted only by authorized personnel.
During inspection it was noted that computers used in the laboratories and production areas had recent files [word and excel] deleted from the system and the “Recycle Bin” was emptied. Repeated false statements were provided by multiple employees before admitting the deletion of files.
Filter integrity test results can be deleted from the Sartorious tester, glove integrity test can be deleted, production supervisor has access to change the data/ time on the system.
Ob9: Data is not documented contemporaneously.
a. Repeat observation – sampling records does not have the sign or date of the record, environmental sampling records are not made at the time of sampling.
b. Swabs used for surface monitoring are not labelled at the time the samples are collected.
c. Batch records have missing entries.
Ob10: Thorough review of documents is not performed.
Documentation of settle plates was done differently by different analyst. Some recorded the start time and end time of the exposure, some mentioned only the total time of the exposure though the reviewer approved both the documents.
Ob11: Procedures for maintenance of equipment had not been established and followed.
Approved procedures for preventive maintenance did not include maintenance recommended by the equipment manufacturer – dust and unidentified black residue was observed over equipment not dedicated to products.
Ob12: Samples collected for finished product release testing is not satisfactorily representative of the manufactured batch.
Repeat observation – No evidence and/ or documentation to support when and how the samples were collected throughout the batch manufacturing process.
Ob13: Samples collected to evaluate conformance of a batch are not representative
This re-inspection and the subsequent Form 483 will set back some of the product launches the company had been banking on for quite some time now.
We can conclude that most of the observations are of medium and high risk in nature. Considering some of these are repeat observations from the initial inspection and subsequent inspection, this facility getting into the good books of the USFDA does not look to happen at the least until the end of this year.
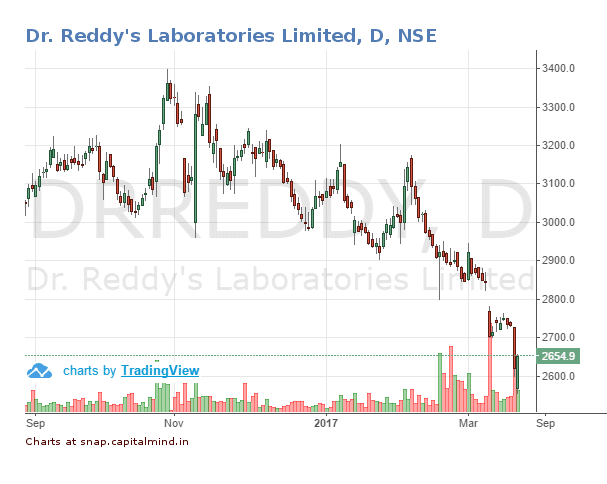
The stock which did not tank much after Miryalaguda Form 483, fell close to 3% and has touched a 52 week low of Rs. 2,650/- yesterday.
Surajit Pal, an analyst at Prabhudas Lilladher said:
This is a negative development as the number of observations is high despite the remedial measures taken by Dr.Reddy’s after receiving the warning letter. Duvvada plant is newer but USFDA is very strict as far as injectables are concerned. I believe that remediation process will take at least one to one-and-a-half years and this will impact future product filings and approvals in the US.
Angel Broking said in a client note:
The site manufactures cytoxic and hormonal injectable and is an important plant given Dr Reddy’s focus on complex generic filling. Since the US contributes nearly half of the company’s sales this could hit the incremental business impacting the near term profitability.
Love it or hate it, do leave us with your reviews in the comment section below!


